

Autofagie is een evolutionair geconserveerd celfenomeen waarbij beschadigde organellen en overtollige eiwitten worden afgebroken om de juiste cellulaire balans te behouden tijdens stress. Het omvat de vorming van dubbelmembraanblaasjes, autophagosomes genaamd, die cytoplasmatische lading vastleggen en naar lysosomen transporteren. Autophagy speelt een cruciale rol bij het handhaven van de cellulaire energie- en fysiologische balans. Ontregeling kan leiden tot ziekten zoals ontsteking, veroudering, stofwisselingsziekten, neurodegeneratieve aandoeningen en kanker. Het begrip van de moleculaire mechanismen en hun rol in ziekteontwikkeling staat nog in de kinderschoenen. Veel farmacologische behandelingen en gezonde levensstijlen richten zich op het herstellen van autofagie, waardoor het een potentieel doel wordt voor de volksgezondheid.
1. Introductie
Deregulatie van autofagie is betrokken bij de pathogenese van een breed scala aan menselijke ziekten. Het is een proces waarbij cellen zichzelf 'eten' om onnodige of beschadigde componenten te elimineren en celhomeostase te handhaven. Autophagy is essentieel voor het handhaven van de energiebalans en het voorkomen van celbeschadiging. Diverse ziekten, waaronder kanker, metabole stoornissen, neurodegeneratieve en inflammatoire aandoeningen, worden geassocieerd met veranderingen in autophagy. Onderzoek heeft specifieke genmutaties gelinkt aan verschillende aandoeningen, waaronder kanker, astma, lupus, ALS, Parkinson, en diabetes. Het begrijpen van de rol van autophagy is cruciaal voor therapeutische strategieën en het overwinnen van ziekten.
2. Communicatie tussen Autofagische, Apoptotische en Necrotische Routes
Autofagie is een proces in je lichaam dat zorgt voor het behoud van de balans binnen de cellen door onnodige eiwitten en beschadigde organellen te verwijderen. Dit proces wordt geactiveerd onder normale omstandigheden (wanneer er een gebrek aan voedingsstoffen is of een tekort aan groeifactoren) of als reactie op verschillende vormen van stress (zoals zuurstoftekort, inductie van oxidatieve stress of blootstelling aan giftige stoffen). Autofagie is belangrijk voor de cel omdat het ervoor zorgt dat de energiebalans behouden blijft, waardoor zelfs kleine veranderingen in energieniveaus geen schade aan de cel kunnen toebrengen. De basisniveaus van autofagie in de cel, dat wil zeggen de niveaus die aanwezig zijn zelfs zonder specifieke stimuli, zorgen voor de normale vernieuwing van eiwitten en organellen door overbodige organellen en niet-noodzakelijke samengeklonterde eiwitten te verwijderen. Dit proces stelt cellen ook in staat om energie te genereren wanneer voedingsstoffen schaars zijn en biedt bio-energetische ondersteuning tijdens de ontwikkeling (overlevingsrespons).
Als autofagie wordt geremd, wordt er een ophoping van beschadigde organellen waargenomen; eiwitaggregatie en een tekort aan de juiste energievoorziening leiden tot celdood. Met een ongecontroleerd autofagisch proces worden vitale celorganellen en nuttige biologische macromoleculen afgebroken, anti-apoptotische factoren worden verteerd, met als gevolg verstoring van de fysiologische overlevingsmechanismen, wat de dood van de cel zelf kan veroorzaken, oftewel autofagische celdood.
Sommige onderzoeken hebben gemeld dat, tijdens de ontwikkeling van de fruitvlieg (Drosophila), autofagie betrokken was als een fysiologisch relevant model van celdood, terwijl apoptose en necrose niet geactiveerd waren [12]. Het is echter belangrijk om voorzichtig te zijn met het gebruik van de term "autofagische celdood". Aangezien autofagie typisch een proces is dat de overleving bevordert, is het noodzakelijk om voldoende bewijs te hebben om te bewijzen dat het een oorzakelijke rol heeft bij celdood [13]. We moeten aantonen dat andere mechanismen van celdood niet verantwoordelijk zijn en controleren of remming van autofagie, door genetische of chemische middelen, celdood voorkomt. Bovendien zijn morfologische criteria alleen niet voldoende om celdood te beoordelen; het is belangrijk om meer dan één test te gebruiken om celdood te meten en vooral om de levensvatbaarheid van de cellen te beoordelen [14].
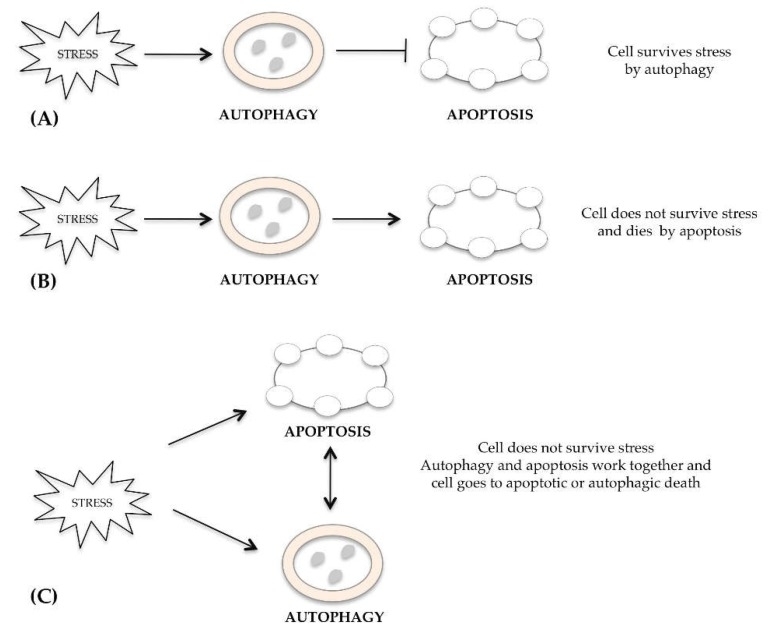
De mechanismen die verschillende vormen van celdood reguleren, zoals apoptose, autofagie en necrose, zijn vaak gescheiden, maar soms overlappen ze en kunnen ze gelijktijdig of achtereenvolgens worden geactiveerd als reactie op een overmatige stimulus van de cel. Veel onderzoekers hebben gewezen op een verband tussen autofagie en apoptose [15]. In dezelfde cel kunnen zowel apoptose- als autofagiepaden tegelijkertijd of na elkaar voorkomen als reactie op dezelfde stimulus, zowel wanneer autofagie beschermend is als wanneer het celdood induceert. Na een stresssignaal activeert de cel overlevingsautofagie die apoptotische celdood remt (Figuur 1A). In andere gevallen kan het autofagische proces de cel niet beschermen tegen de schade veroorzaakt door stress en sterft de cel door apoptose (Figuur 1B). Alternatief werken autofagie en apoptose samen en sterft de cel door beide mechanismen (Figuur 1C). De subcellulaire compartimenten waar autofagie en apoptose met elkaar kunnen worden verbonden, zijn het endoplasmatisch reticulum [16], mitochondriën [17] en lysosomen [18].
3. Moleculaire Regulatie van Autofagie
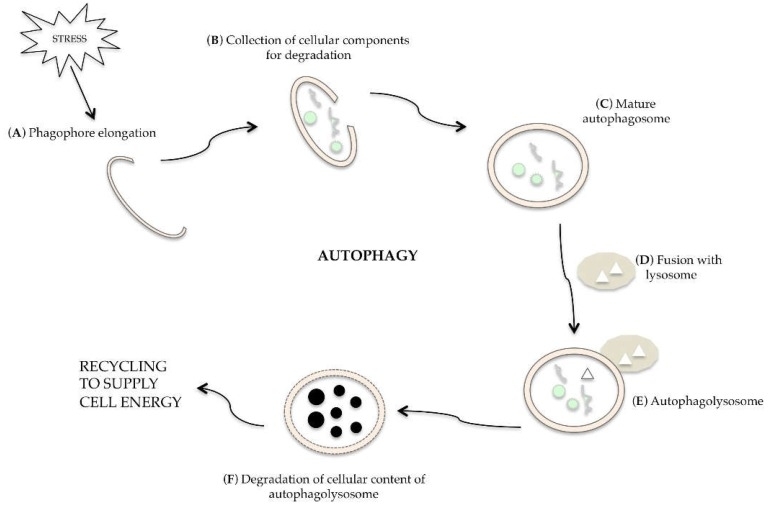
Autophagy is een dynamisch proces waarbij een dubbelmembraan (phagophore) ontstaat, organellen en macromoleculen omwikkelt om een autophagosome te vormen. Dit fusioneert met een lysosoom tot een autophagolysosoom, waar lysosomale enzymen de inhoud afbreken. Vrijgekomen macromoleculen kunnen worden gebruikt voor biosynthese of celenergie. Het proces is cruciaal voor celoverleving en kan worden gereguleerd door verschillende signaalroutes. Autophagy kruist soms met apoptose, waarbij autophagy celoverleving kan bevorderen of leiden tot celvernietiging. Begrip van deze processen is van belang voor ziektebehandeling.
Genetische tests op de gist Saccharomyces cerevisiae hebben geholpen bij het begrijpen van de moleculaire regulatie van autophagy door de identificatie van ongeveer 30 genen, de zogenaamde ATG-genen (AuTophaGy gerelateerde genen). Deze genen reguleren autophagy en hebben homologe sequenties in hogere eukaryoten, van fruitvlieg tot zoogdieren, wat suggereert dat het moleculaire mechanisme van autophagy sterk geconserveerd is in de evolutie.
Bij voldoende voedingsstoffen remmen de mammaliaanse doelwitten van rapamycine (mTOR) en proteïnekinase A (PKA) autophagy negatief door de fosforylering en remming van het inductiecomplex, het unc-51-achtige kinase 1 (ULK1)-complex. Bij schaarste aan voedingsstoffen worden verschillende chemische mediatoren vrijgegeven als reactie op stress, zoals een stijging van de AMP-ATP-verhouding, verhoogde productie van reactieve zuurstofspecies (ROS), toename van nicotinamide-adeninedinucleotide (NAD+), remming van mTOR-activiteit en activering van AMP-geactiveerde proteïnekinase (AMPK), waardoor de autophagische machine wordt gestart. De inductiefase van phagophore-vorming wordt bemiddeld door het ULK1-complex, bestaande uit ULK1, ATG13, focal adhesion kinase family interacting protein van 200 kDa (FIP200) en ATG101-proteïne. Het geactiveerde ULK1-complex werkt op het complex dat autophagosome-nucleatie reguleert en activeert het fosfatidylinositol 3-kinase-complex (PtdIns3K). Dit complex, bestaande uit Beclin1, ATG14, vacuolar protein sorting (VPS)15, VPS34, activating molecule in BECN1 regulated autophagy protein 1 (AMBRA1) en ultraviolet irradiation resistance-associated gene (UVRAG), genereert het lipide fosfatidylinositol-3-fosfaat (PI3P). PI3P rekruteert andere essentiële eiwitten voor de vorming van vacuolen, zoals het WD-repeat protein interacting with phosphoinositides (WIPI) -eiwit.
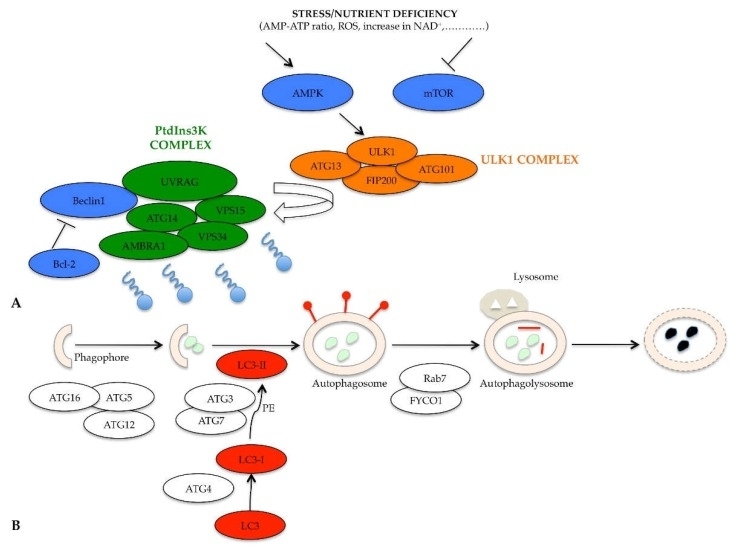
Naast de remming van mTOR en activatie van AMPK wordt de activiteit van het PtdIns3K-complex verder gereguleerd door het Beclin1-Bcl-2-complex. Bij overvloedige voedingsstoffen vormt het Beclin1-eiwit een complex met het Bcl-2-eiwit, dat Beclin1-recruitment en -functie in het PtdIns3K-complex remt. Bij voedingsstoffentekort wordt het Beclin1-Bcl-2-complex gedissocieerd en kan Beclin1 vrij functioneren. De fasen van phagophore-vorming en autophagosome-regulatie worden voornamelijk beheerst door het ATG12-ATG5-ATG16-complex, essentieel voor de verlenging van het membraan tot de vorming van phagophore. Een transmembraanproteïnesysteem met ATG9, ATG2 en WIPI 1/2 is ook nodig voor de verlenging van de phagophore-structuur. Het tweede conjugatiecomplex, essentieel voor de voortgang van het proces, is het ATG8-eiwit, geïdentificeerd als microtubule-associated protein 1 light chain 3 (MAP1-LC3 of kortweg LC3) bij zoogdieren. De volledige autophagosome begint zijn reis langs het microtubule naar het lysosoom, waarmee fusie plaatsvindt. Het transport langs het microtubule wordt bemiddeld door een adaptief eiwitcomplex gevormd door LC3, Rab7 en FYCO1. Na de vorming van autophagolysosoom wordt het LC3-II-eiwit geïnternaliseerd, wordt het PE-residu verwijderd dankzij de werking van lysosomale enzymen en wordt het eiwit vrijgegeven in het cytoplasma, met een daaropvolgende afname van de expressie.
4. Selectieve Autophagy
Cellulaire energie, nodig om de ongunstige omstandigheden door het ontbreken van voedingsstoffen te overleven, komt voort uit de niet-selectieve afbraak van beschadigde cellulaire componenten. In andere situaties, zoals stress die het endoplasmatisch reticulum beschadigt of veranderingen in het celmetabolisme, identificeert het autophagische apparaat specifiek het te elimineren cytoplasmatische organel en elimineert het via een mechanisme genaamd "selectieve autophagy". Er zijn verschillende soorten selectieve autophagy, afhankelijk van de geëlimineerde organel: mitophagy (voor mitochondriën), ribophagy (voor ribosomen), reticulophagy (voor het endoplasmatisch reticulum), lysophagy (voor lysosomen), pexophagy (voor peroxisomen), lipophagy (voor lipidedruppels), glycophagy (voor glycogeen), aggrephagy (misvormde eiwitten) en xenophagy (geïnfecteerde pathogenen).
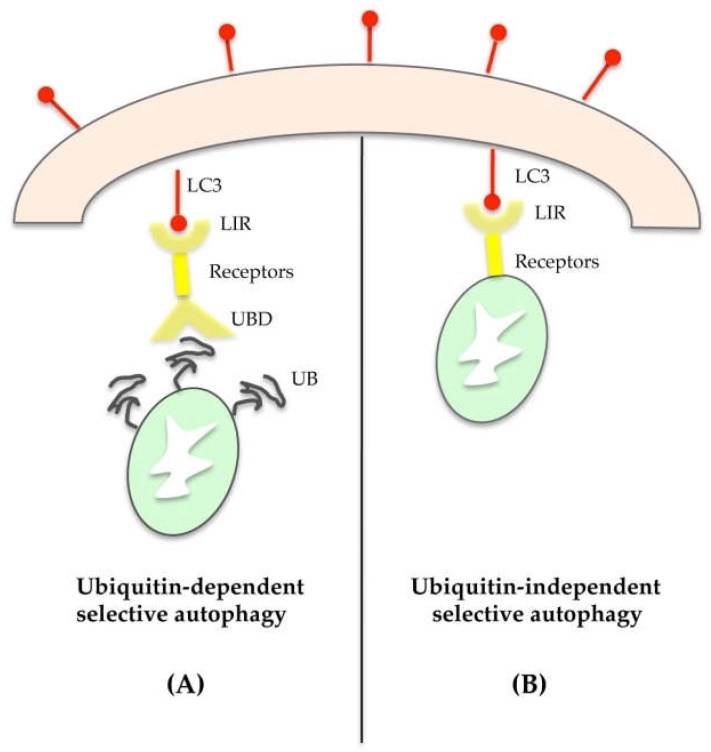
Organel kan selectief worden afgebroken via ubiquitine-afhankelijke of -onafhankelijke mechanismen. In het eerste geval heeft het doelorganel een polyubiquitineketen die het naar substraatspecifieke receptoren leidt, zoals p62, de buur van het BRCA1-gen 1 (NBR1), nucleair domein 10-eiwit 52 (NDP52) en optineurine. Deze eiwitten fungeren als moleculaire adapters omdat ze in staat zijn om aan de ene kant de polyubiquitineketen (UB) op beschadigde organellen te herkennen en te binden via de UB-bindingdomein (UBD), en aan de andere kant het LC3-II-eiwit in het autophagosome te binden via LC3-interagerende regio's (LIR) (Figuur 4A). Dus, naast zijn rol in de expansie van het autophagosome, speelt LC3-II ook een essentiële rol in de selectie van lading.
p62, een 62 kDa-eiwit ook bekend als sequestosome 1 (SQSTM1), bevindt zich doorgaans op geubiquitineerde eiwitten en wordt in autophagosome-vacuolen geïsoleerd. Het is wijdverspreid tot expressie gebracht in weefsels zoals het zenuwstelsel, endocriene, voortplantings- en immuunsysteem, waar het een belangrijke rol speelt bij de pathogenese van ziekten. p62, als het belangrijkste selectieve substraat, wordt gerekruteerd naar voorbestaande isolatiemembranen via interactie met LC3. Extra studies tonen aan dat p62 zich lokaliseert naar de autophagosome-vormingsplaats op het endoplasmatisch reticulum zonder interactie met LC3 of andere ATG-eiwitten. Vanwege zijn rol bij het leveren van autofagische lading, is de expressie van p62 omgekeerd gecorreleerd met autofagische degradatie en kan het worden gebruikt als een maat voor autofagische flux.
Een ander ladingsreceptor is NBR1, die verschillende domeinen en kenmerken deelt met p62, zoals het C-terminale domein dat polyubiquitineketens bindt, en de sequentie die interageert met ATG8. Onlangs is aangetoond dat NBR1 en p62 samenwerken om selectief polyubiquitine-eiwitaggregaten naar autofagosomen te richten en peroxisomen af te breken via pexophagy.
Wat betreft het ubiquitine-onafhankelijke mechanisme kunnen talrijke autofagische receptoren op organellen hun lading rechtstreeks verbinden met het autofagosoom (Figuur 4B). Als voorbeeld kan mitophagy Bcl-2/adenovirus E1B 19-kDa interactie-eiwit 3 (BNIP3) of BNIP3-achtig (bekend als NIX) omvatten, die rechtstreeks interageren met het LC3-II van het autofagosoom.
Hoewel niet-selectieve autophagy essentieel is voor cellevitaliteit, speelt selectieve autophagy een directe rol bij een breed scala aan ziekten.
5. Detectiemethoden van Autophagy in Zoogdiercellen
Autophagy is een zeer complex en dynamisch proces dat moeilijk te analyseren kan zijn in in vitro of in vivo modellen. Veel onderzoekers die de afgelopen jaren complexe studies over autophagy hebben uitgevoerd, hebben technische en wetenschappelijke bijdragen geleverd om een uitgebreide publicatie op te stellen voor het interpreteren van monitoring-autofagie-assays. Hieronder worden geschikte technieken voor in vitro autophagy-onderzoek, technische maatregelen om de vorming van verwarrende artefacten te voorkomen, en richtlijnen voor juiste gegevensinterpretatie geïllustreerd. De belangrijkste in vitro methoden voor het monitoren van autophagy worden samengevat in Tabel 2.
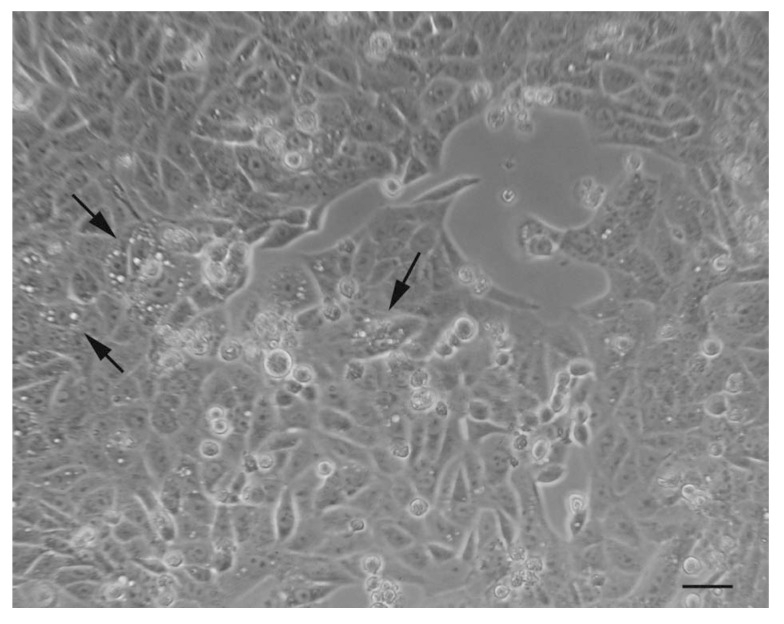
De eerste waarnemingen van autophagische monsters onder een fase-contrast optische microscoop leveren een choreografisch beeld van het fenomeen, waarbij de aanwezigheid van "gaten" wordt benadrukt, die niets anders zijn dan zeer grote vacuolen binnenin het cytoplasma (Figuur 5).
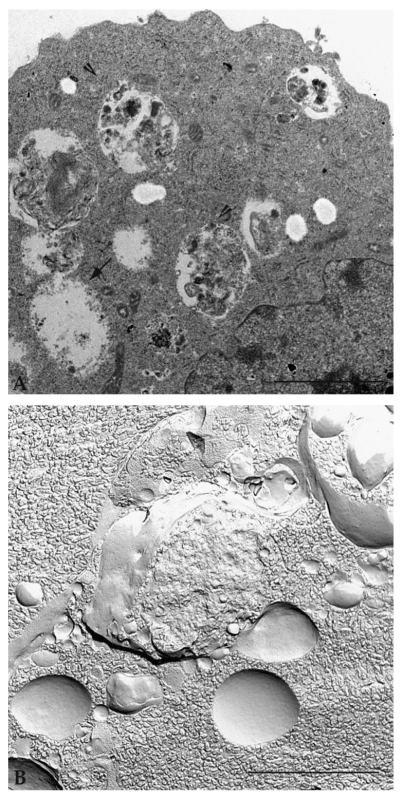
Gedetailleerde analyse van hetzelfde monster met een transmissie-elektronenmicroscoop toont cytoplasmatische vacuolen met enkele en dubbele membranen, bevattende cytoplasmatische organellen en materiaal dat moet worden verteerd en gerecycled (Figuur 6A). Observatie van monsters die zijn voorbereid volgens de standaard inclusieprocedures maakt het mogelijk de verschillende fasen van de vorming en rijping van het autophagische vacuool (initiatie, nucleatie, fagoforevorming, autofagosoom, fusie van autofagosoom met lysosoom en autofagolysosoom) te definiëren [53]. Als de monsters worden voorbereid met behulp van een snelle invriestechniek met plasmamembraanbreuk (bekend als de freeze-fracturingtechniek), wordt een diepgaand begrip van de vacuolaire moleculaire membraanorganisatie verkregen (Figuur 6B).
Autofagosomen kunnen ook worden herkend met behulp van een specifieke marker die ze identificeert. Sommige karakteristieke eiwitten van het autofagische pad kunnen worden geïdentificeerd met behulp van fluorescerende antilichamen specifiek voor een eiwit, en door de monsters te observeren met de fluorescentiemicroscoop. De veranderingen die zowel in de lokalisatie van het LC3-eiwit als in de moleculaire omzetting worden waargenomen, zoals de omzetting van LC3-I naar LC3-II, kunnen worden gekwantificeerd door het autofagie-mechanisme te stimuleren of te remmen met selectieve geneesmiddelen via de western blot-techniek. Om de dubbele rol van autofagie als mechanisme voor celoverleving of celdood te verduidelijken, zijn interessante resultaten verkregen door ATG-genen te dempen. Deze genen zijn autofagiecontrollers, dus eenmaal het interessante gen is gedempt, is het mogelijk om te evalueren hoe de expressie van pro-autofagische of pro-apoptotische eiwitten varieert. Deze resultaten zullen ons helpen begrijpen of autofagie betrokken is bij het onderzochte biologische proces met het oog op celoverleving of celdood [54].
Uit deze snelle beschrijving van technieken is het gemakkelijk te begrijpen dat de autofagie-mechanismen zeer dynamisch en complex zijn, met name wanneer het onderzoek zich moet onderscheiden tussen autofagische celoverleving of celdood, of tussen autofagie en apoptose. Om beoordelingsfouten te voorkomen, wordt het gelijktijdige gebruik van meerdere tests aanbevolen om zeker te zijn van de uitgevoerde evaluatie.
6. Autofagie, Ontsteking en Veroudering
Autofagie is geïdentificeerd als de belangrijkste regulator van het inflammasoom; een belangrijk aangeboren immuunpad geactiveerd door exogene stimuli, zoals pathogene micro-organismen, of door endogene mediatoren, zoals ROS, mitochondriale schade en omgevingsirritanten [55]. Inflammasoomactivatie omvat de vorming en oligomerisatie van een eiwitcomplex, waaronder een nucleotide-oligomerisatiedomein (NOD)-achtige receptor (NLR), een adapter-eiwit en pro-caspase-1. Deze activatie zorgt voor de splitsing en activering van caspase-1, gevolgd door de afgifte van pro-inflammatoire cytokines, zoals interleukine (IL)-1β en IL-18, uit aangeboren immuuncellen [56]. In het bijzonder, wanneer endogene mediatoren een massale inflammatoire respons veroorzaken, kunnen ze weefselschade veroorzaken en de ontwikkeling van inflammatoire ziekten bevorderen. Daarom is negatieve of positieve regulatie van het inflammasoom essentieel om een goede gezondheidstoestand te waarborgen.
Zoals aangetoond door meerdere studies, kan autofagie inflammasoomactivatie negatief reguleren via verschillende mechanismen:
1. Door het verwijderen van beschadigde organellen zoals mitochondriën, wat leidt tot verminderde afgifte van ROS en daaropvolgende onderdrukking van inflammasoomactivatie.
2. Door p62-afhankelijke afbraak van inflammasoomcomplexen en mitochondriën.
3. Door pro-IL-1β in autofagosomen te sequestreren voor afbraak. Dit pad draagt bij aan autofagie-gemedieerde vermindering van IL-1β-secretie [57,58,59].
Een tekort aan autofagie veroorzaakt inflammatoire ziekten gerelateerd aan het inflammasoom; genetische studies hebben aangetoond dat polymorfismen van het ATG16L1-gen geassocieerd zouden kunnen zijn met de ziekte van Crohn, een chronische inflammatoire darmaandoening [60,61,62]. Helaas is de pathofysiologie van de ziekte van Crohn nog in onderzoek; in feite hebben sommige auteurs [63,64] aangetoond dat andere genen en predisponerende omgevingsfactoren de ontwikkeling van de ziekte bij patiënten kunnen beïnvloeden [65,66]. Daarom moet voorzichtigheid worden betracht om het werkelijke belang van autofagie in inflammatoire darmaandoeningen te onderscheiden.
Over het algemeen suggereren deze gegevens dat inflammasoom en autofagie elkaar wederzijds reguleren, waarbij ze het evenwicht bevorderen tussen ontstekingsrespons om zichzelf te verdedigen tegen de gastheer en het voorkomen van een overmatige ontstekingsrespons die weefselschade en inflammatoire ziekten kan veroorzaken [67].
Recente studies hebben aangetoond dat de verminderde activiteit van autofagie die veroudering kenmerkt, te wijten is aan de ophoping van disfunctionele mitochondriën, ROS en NLRP3 inflammasoomactivatie in macrofagen [68]. Deze factoren maken cellen vatbaarder voor verouderingsgerelateerde ziekten, zoals atherosclerose en type 2 diabetes. In het bijzonder zijn de activiteiten van drie verouderingsgerelateerde signaalroutes geïdentificeerd: lagere activiteit van insuline/insuline-achtige groeifactor 1 (IGF-1) signalering, mTOR en Sirtuin-1 (Sirt-1) netwerk. Voor verouderingsgerelateerde ziekten is het raadzaam om een dieet te volgen dat rijk is aan voedingsmiddelen met natuurlijke anti-verouderingsverbindingen zoals resveratrol, catechinen, epigallocatechine-3-gallaat, luteoloside of propolis [69]. Recente studies hebben aangetoond dat resveratrol, een natuurlijk polyfenol in rode wijn, NAD+-afhankelijke deacetylasen zoals Sirt-1 activeerde, wat een veelbelovend therapeutisch doelwit is voor verouderingsgerelateerde ziekten [70]. Bovendien kon dit natuurlijke product autofagie-activatie stimuleren door NLRP3-activatie te onderdrukken [71].
7. Rol van Autofagie bij Ziekten van het Metabool Syndroom
De rol van autofagie bij het metabool syndroom is bijzonder interessant. Het metabool syndroom, zoals gedefinieerd door de NHS, is een multifactoriële ziekte, vooral in geïndustrialiseerde landen, en wordt gekenmerkt door ten minste drie van deze risicofactoren:
1. Hoge bloeddruk (boven 85/130 mmHg).
2. Hoge triglyceriden (boven 150 mg/dL).
3. Lage niveaus van high-density lipoprotein (HDL)-cholesterol (lager dan 40/50 mg/dL).
4. Hoge bloedsuikerspiegel (boven 100 mg/dL).
5. Viscerale verdeling van lichaamsvet [72].
De gelijktijdige aanwezigheid van deze factoren stelt de patiënt bloot aan het potentiële gevaar van het ontwikkelen van type 2 diabetes, obesitas en bijbehorende hartziekten. Het begin van deze pathologie wordt vaak begunstigd door familiaire aanleg en een ongezonde levensstijl, gekenmerkt door een incorrect en onevenwichtig dieet en onvoldoende lichaamsbeweging. Om de rol van autofagie bij het metabool syndroom te begrijpen, moet men stilstaan bij het fysiologische defect dat type 2 diabetes definieert. Type 2 diabetes wordt gekenmerkt, enerzijds, door een tekort aan insulineafscheiding en, anderzijds, door insulineresistentie (de geproduceerde insuline werkt niet bevredigend op de doelorganen). De gecombineerde werking van deze twee processen is verantwoordelijk voor hyperglycemie, kenmerkend voor type 2 diabetes (ook wel diabetes mellitus genoemd). De toename van de bloedglucoselevels veroorzaakt, op systemisch niveau, in de cellen van de doelorganen, de volgende disfuncties: toename van reactieve zuurstofspecies en afname van antioxidatiesystemen (activering van oxidatieve stress); mitochondriale disfuncties; en activering van het ontstekingsproces, wat leidt tot nefropathie, voetzweren, beroerte, neuropathie, retinopathie en hartziekte (Figuur 7).
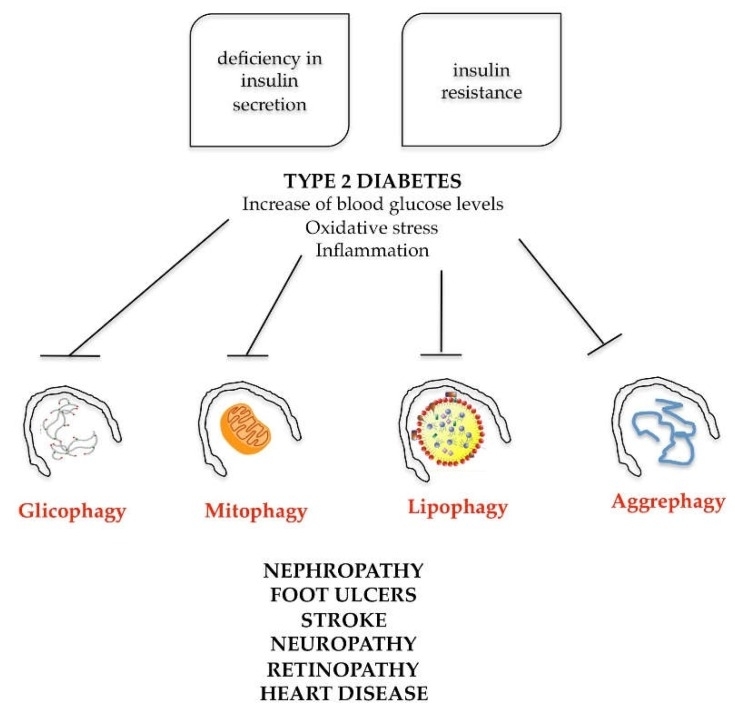
De voortgang van type 2 diabetes wordt geassocieerd met autofagie via insulineafscheidingsdeficiëntie en de ontwikkeling van insulineresistentie [73]. Autofagie heeft een beschermende rol tegen apoptotische celdood van pancreatische bètacellen, waarbij de structuur en functie behouden blijven. Bovendien beschermen selectieve autofagische mechanismen, zoals lipofagie, aggrephagie, mitofagie en glycofagie, de doelorganen van insuline (lever, vetweefsel, skeletspieren, nieren) tegen door hyperglycemie veroorzaakte schade door oxidatieve stress. Dientengevolge veroorzaken meerdere veranderingen in autofagie-mechanismen verstoring van het metabolisme en de cellulaire balans, wat de ontwikkeling van pathologische situaties zoals diabetes, obesitas en cardiovasculaire complicaties bevordert [74].
Het beschermende effect van autofagie op metabool actieve organen is wetenschappelijk aangetoond door dierstudies. In de lever bijvoorbeeld, zorgt autofagie voor de juiste functie van hepatocyten en regelmaat in het metabolisme van glycogeen en triglyceriden. Studie van diermodellen waarbij hepatocyten de verwijdering van het ATG7-gen ondergingen, toont een ophoping van veranderde en vervormde mitochondriën, van lipide druppels, en een toegenomen aantal geübiquitineerde eiwitaggregaten [75]. Op het niveau van de alvleesklier zorgt autofagie voor het behoud van de structuur, massa en functionaliteit van bètacellen en vermindert het de stressniveaus die worden geproduceerd door verstoringen van het endoplasmatisch reticulum [76]. In vetweefsel zorgt autofagie voor de differentiatie van adipocyten: het verwijderen van de ATG5- en ATG7-genen in adipocyten veroorzaakt abnormale ophoping van lipiden en abnormale transformatie van wit vetweefsel in bruin vetweefsel [77].
Eindelijk, in skeletspieren zorgt autofagie voor een grotere tolerantie voor glucose en een goede instandhouding van de spiermassa zelf. De verwijdering van de ATG5- en ATG7-genen in de skeletspieren veroorzaakt bij muizen verlies van spiermassa en vetmassa, en ook schade op het niveau van het vetweefsel (omzetting van wit vetweefsel naar bruin en grotere beta-oxidatie) [78]. Een recente review vat studies samen die verband houden met de rol van multifunctioneel p62 bij metabole ziekten, waaruit blijkt dat p62 een potentieel doelwit zou kunnen zijn. Het p62-eiwit beschermt de bètacellen van de alvleesklier tegen apoptose en vermindert insulineresistentie; het controleert de eiwitkwaliteit van cardiomyocyten door autofagie-activatie en speelt een beschermende rol bij het verminderen van de ontwikkeling van diabetische nefropathie door de activatie van de epidermale groeifactorreceptor (EGFR) te remmen [79].
Gezien de centrale rol van autofagie bij het metabool syndroom, is de beste therapeutische strategie om deze ziekte te bestrijden het gebruik van autofagie als doelwit via calorierestrictie en intermitterend vasten, lichaamsbeweging en, indien nodig, farmacologische behandeling (Figuur 8) [80].
Figuur 8 geeft een schematische voorstelling van de regulatie van autofagie door vasten en calorierestrictie, lichaamsbeweging of farmacologische behandeling. (A) Vasten en calorierestrictie werken op Sirt-1 en HSP70-eiwitten met autofagische en anti-apoptotische effecten, respectievelijk; (B) Lichaamsbeweging induceert autofagie, verstoort het Beclin1-Bcl-2-complex en balanceert mitochondriale fusie/fissie; (C) Behandelingen met Metformine of natuurlijk extract van Moringa oleifera herstellen mitofagie en lipofagie.

Vasten en calorierestrictie hebben gunstige effecten op de menselijke gezondheid [81], omdat ze in staat zijn om de fysiologische niveaus van autofagie te herstellen door de activering van Sirt-1 en AMPK (Figuur 8A) [82,83]. Sirt-1 is een NAD+-afhankelijke deacetylase die betrokken is bij glucosemetabolisme en insulinesecretie; dit eiwit wordt geremd in cellen met een hoge resistentie tegen insuline [84]. Het functioneert als een metabole sensor die de toename van NAD+-concentraties detecteert als gevolg van verbeterde NADH-oxidatie. Eenmaal geactiveerd door calorierestrictie kan Sirt-1 essentiële autofagische modulatoren, zoals ATG5 en ATG7, deacetyleren, waardoor de autofagische effecten worden gestimuleerd [85]. Bovendien zijn vasten en calorierestrictie in staat om vasculaire disfunctie en cardiovasculair risico geassocieerd met metabole ziekten te verminderen door modulatie van ontstekingscytokines en oxidatieve stress [86]. In het bijzonder verhoogt vasten de transcriptie van stressgeïnduceerde eiwitten, zoals heat shock protein 70 (HSP70), dat anti-inflammatoire en anti-apoptotische eigenschappen heeft. In de skeletspieren van diabetische patiënten nemen de niveaus van HSP70 af, wat gerelateerd is aan insulineresistentie; dus verhogingen van het HSP70-niveau door vasten verzachten insulineresistentie en bijbehorende vasculaire disfunctie [87].
De gunstige effecten van lichaamsbeweging bij mensen met type 2 diabetes en cardiovasculaire complicaties zijn goed gedocumenteerd. Lichaamsbeweging activeert een cascade van gebeurtenissen die leidt tot een lichte toename van oxidatieve stress, energieonevenwicht, intracellulaire calciumniveaus en de remodeling van eiwitten. Al deze gebeurtenissen activeren het overlevingsmechanisme van autofagie, dat de omzet van mitochondriën en eiwitten vergemakkelijkt en de stofwisselingsmodificatie. Deze aanpassingsreacties leiden over het algemeen tot optimalisatie van lipide- en glucosehomeostase en verbetering van prestaties van weerstand [88]. In het bijzonder werkt lichaamsbeweging in skeletspiercellen door het Beclin1-Bcl-2-complex te vernietigen, dat essentieel is voor de opstartfase van autofagie (Figuur 8B) [89].
Lichaamsbeweging speelt een gunstige rol bij obese individuen. In de cellen van obese personen kunnen defecten in de mitofagie leiden tot oxidatieve stress, ophoping en disfunctie van mitochondriën en verstoring van het evenwicht tussen fission- en fusieprocessen in mitochondria (fundamenteel voor het behoud van een goede staat van mitochondriale gezondheid). Hoewel moleculaire en cellulaire mechanismen niet duidelijk zijn opgehelderd, grijpt lichaamsbeweging in door de fysiologische niveaus van mitofagie en de fusie- en fissieprocessen te herstellen, waardoor apoptotische gebeurtenissen in obese skeletspieren worden verminderd [90].
Wat betreft farmacologische interventies worden diabetische patiënten behandeld met metformine, een hypoglycemisch medicijn met veel bijwerkingen. Metformine, werkzaam op het AMPK-eiwit, is in staat zowel mytofagie als lipofagie te activeren en de activering van het inflammasoom te blokkeren [91]. Gezien de bijwerkingen van metformine en het optreden van geneesmiddelresistentie, gezien de nieuwe kennis met betrekking tot de moleculaire paden die autofagie reguleren, evolueert het onderzoek voortdurend naar natuurlijke stoffen met
hypoglycemische effecten [92]. Recente studies hebben aangetoond dat extracten van de Moringa oleifera-plant, rijk aan flavonoïden, isothiocyanaten en polyfenolen, meerdere functies vertonen, waaronder ontstekingsremmende, hypoglycemische en bloedlipidenverlagende functies [93]. De moleculaire mechanismen die verantwoordelijk zijn voor deze multifunctionaliteit zijn de inactivatie van NF-kB- en PI3K/AKT-pathways en de modulatie van beschermende actie op de mitochondriale ademhalingsketen gemedieerd door Sirt-1 (Figuur 8C) [94].
8. Autofagie en neurodegeneratieve aandoeningen
De aggregatie van verkeerd gevouwen eiwitten en het verlies van bepaalde neuronale populaties zijn kenmerkend voor de expressie van pathologische neurodegeneratieve ziekten zoals de ziekte van Alzheimer, de ziekte van Parkinson, de ziekte van Huntington en amyotrofe laterale sclerose. Autofagie is gemeld betrokken te zijn bij het optreden van neurodegeneratieve aandoeningen, als het belangrijkste intracellulaire systeem voor degradatie van beschadigde organellen en geaggregeerde eiwitten. Bij neurodegeneratieve ziekten is een verstoring van het rijpingsmechanisme van de autofagosoom in de autofagolysosoom gevonden.
Het is aangetoond dat verwijdering van de ATG5- en ATG7-genen neurodegeneratie veroorzaakt in het centrale zenuwstelsel van muizen [95].
Bovendien speelt autofagie een belangrijke rol bij de afbraak van verschillende eiwitten die verband houden met degeneratieve ziekten, zoals gemuteerd α-synucleïne bij de ziekte van Parkinson, gemuteerd huntingtine bij de ziekte van Huntington, en het gemuteerde TAR DNA-bindende eiwit 43 (TDP-43) bij amyotrofe laterale sclerose.
Bij de ziekte van Alzheimer is de aanwezigheid van extracellulaire amyloïd-β-plaques en intracellulaire neurofibrillaire kluwens, bestaande uit hypergefosforyleerde tau-eiwitaggregaten, onthuld [96]. In de gezonde hersenen zijn de autofagosoomblaasjes niet erg zichtbaar; daarentegen zijn in de hersenen van mensen met de ziekte van Alzheimer talrijke autofagosomen merkbaar. De ophoping van autofagie vacuolen komt voort uit verminderde klaring in plaats van inductie van autofagie, wat suggereert dat de late stadia van autofagie-regulatie een mogelijke therapeutische strategie kunnen zijn voor de ziekte van Alzheimer. Het transmembraaneiwit betrokken bij de fusie van autofagosoom en lysosoom is preseniline 1 (PS1). Dit gemuteerde eiwit wordt beschouwd als een belangrijke factor bij de inductie van de ziekte van Alzheimer; in feite voorkomt het verlies van fosforylering van PS1 Ser367 de fusie van twee vacuolen [97]. Verdere studies tonen aan dat PS1 de Ca2+ homeostase handhaaft door de lysosoomacidificatie te reguleren [98].
De vermindering van acidificatie verstoort de functionaliteit van de lysosoom, waardoor de fusie met de autofagosoom wordt voorkomen. Deze verstoring leidt tot neuron-dystrofie vergelijkbaar met die bij de ziekte van Alzheimer; uit deze waarnemingen begrijpen we hoe dit organel en de juiste functionaliteit ervan belangrijk zijn voor de verwijdering van geaggregeerde eiwitten.
Andere eiwitten zijn geassocieerd met de ziekte van Alzheimer, zoals de clathrinegroep die bindt aan het fosfatidylinositol-eiwit, dat betrokken is bij vesiculair transport, dat endocytose reguleert en de aggregatie van tau-eiwitten en Beclin1 voorkomt. Beclin1 heeft een sleutelrol bij de vorming van de autofagosoom; caspase 3, een belangrijk activerend component van apoptose, kan het Beclin1-eiwit scheiden en de voortgang van het autofagieproces stoppen.
Het is bekend dat sommige genetische en biochemische defecten de ziekte van Alzheimer kunnen initiëren, waardoor oxidatieve stress en chronische ontsteking ontstaan. In dit verband lijken de toename van antioxidantensystemen en de overeenkomstige enzymniveaus gunstige effecten te hebben op neurodegeneratieve ziekten in diermodellen. Een nucleaire factor die betrokken is bij de pathologie van de ziekte van Alzheimer en geactiveerd wordt als reactie op oxidatieve stress is de nucleaire factor afgeleid van erythroïde 2 zoals 2 (Nrf2) [99]. Deze factor wordt verondersteld autofagie te activeren en geaggregeerde tau-eiwitten te elimineren, via de autofagische receptor NDP52 [100,101].
De ziekte van Parkinson is een veelvoorkomende neurodegeneratieve ziekte en wordt gekenmerkt door een ernstig verlies van dopaminerge neuronen in de substantia nigra pars compacta, gevolgd door polyubiquitinatie en insluiting van α-synucleïne-eiwit, zogenaamde Lewy-lichamen. Deze lichamen lijken kenmerkend te zijn voor de ziekte van Parkinson, maar ook voor andere neurologische aandoeningen [102].
De rol van autofagie bij de ziekte van Parkinson is aangetoond door de aanwezigheid van veranderingen in lysosomen en autofagosomen in neuronen; ter ondersteuning van dit bewijs, wanneer de lysosoom functioneel is aangetast, is de hoeveelheid α-synucleïne verhoogd, wat wijst op een verstoring van het autofagiepad. Het is aangetoond dat een autosomaal recessieve mutatie in het GBA-gen, dat codeert voor lysosomale hydrolase, veranderingen induceert in het autofagosoom-lysosoompad en aggregatie van α-synucleïne [103].
De transcriptiefactor EB (TFEB) is geïdentificeerd als de factor die genen positief reguleert die betrokken zijn bij de vorming van autofagosomen en de fusie van lysosomen, waardoor de klaring van lysosomale exocytose toeneemt [104]. Onlangs is aangetoond dat overexpressie ervan lysosoomschade kan verminderen en daardoor neurologische aandoeningen gerelateerd aan α-synucleïne kan verbeteren.
De gebruikelijke oorzaken van autosomaal dominante vormen van de ziekte van Parkinson zijn te wijten aan mutaties in leucine-rich repeat kinase 2 (LRRK2) en in VPS35 D620N. Bij LRRK2 is aangetoond dat overexpressie de autofagiestroom verandert, terwijl de mutatie van VPS35 D620N het transport van het ATG9-eiwit verandert via de wijziging van het WASH-complex. Autosomale recessieve vormen van de ziekte van Parkinson worden veroorzaakt door mutaties in Parkin RBR E3 ubiquitine-eiwitligase (PARK2) en PTEN-geïnduceerde veronderstelde kinase 1 (PINK1), die de afbraak van beschadigde mitochondriën (mitofagie) reguleert. PINK1 fungeert als een upstream factor, hoopt zich specifiek op in gedepolariseerde mitochondriën, terwijl PARK2 de overdracht van ubiquitine naar mitochondriale substraten katalyseert [105,106].
Huntington's ziekte is een neurodegeneratieve aandoening veroorzaakt door GAG-trinucleotide-herhalingen in het gen dat codeert voor het huntingtine-eiwit (HTT). Huntingtine speelt een belangrijke rol in het transport van autofagie-eiwitten. Depletie van HTT veroorzaakt een abnormale ophoping van autofagosomen met ingesloten mitochondriën. In de ziekte van Huntington kan een polymorfisme in het ATG7-gen, een wijziging in zowel Beclin1 als in het selectieve substraat p62/SQSTM1 van autofagie, worden waargenomen. Dysregulatie van deze systemen is ook zeer belangrijk bij het vroeg begin van de ziekte van Huntington. Daarentegen kan niet-gemuteerd HTT binden aan p62, interageren met ULK1 en autofagie induceren [107].
Amyotrofe laterale sclerose is een neuromusculaire ziekte waarbij progressieve degeneratie van ruggenmerg- en hersenmotorneuronen optreedt tot atrofie en spierverlamming. Tot nu toe zijn ongeveer 30 genen geïmpliceerd, waaronder die betrokken zijn bij autofagie en mitofagie. Er wordt met name een verstoring van de klaring van eiwitaggregaten en beschadigde mitochondriën waargenomen. De genen die zijn geassocieerd met amyotrofe laterale sclerose coderen voor eiwitten die RNA binden, zoals TDP43 en FUS, wat aangeeft dat ribostase-veranderingen de ziekte kunnen bevorderen. Bij het begin van de ziekte zijn ook verstoringen van protease, mitochondriale functie, cytoskeletintegriteit en intracellulair verkeer betrokken. De genen bij amyotrofe laterale sclerose die betrokken zijn bij autofagie zijn SQSTM1, TBK1 en OPTN. Aan de andere kant kunnen genen die vesiculair verkeer reguleren, zoals C9ORF72, VCP, CHMP2B, VAPB, ALS2, DCTN1, ook direct en indirect betrokken zijn bij het autofagie-mechanisme. Bij amyotrofe laterale sclerose worden vaak motorneuronen van patiënten, accumulatie van eiwitaggregaten en opgezwollen of dystrofische mitochondriën aangetroffen. Er is momenteel geen genezing voor deze ziekte [108].
9. Pro-survival autofagie bij kanker
De rol van autofagie bij kanker is controversieel en complex en hangt af van het kankertype, het stadium en de genetische context [10]. Tijdens de vroege fase van kankerontwikkeling beschermt autofagie cellen tegen door ROS veroorzaakte schade aan DNA en eiwitten, waardoor de transformatie van normale cellen naar tumorcellen vertraagt [109]. Het is bevestigd dat muizen met mono-allelische verwijdering van Beclin1 een verhoogde gevoeligheid hebben voor spontane tumorentwikkeling [110].
Tijdens de late stadia, zoals promotie, progressie en metastase, heeft autofagie een pro-tumor effect, omdat het producten van door ROS veroorzaakte metabole stress elimineert en voedingsstoffen levert die nodig zijn voor de overleving van kankercellen [111].
De rol van autofagie is bijzonder interessant bij kankerstamcellen (CSC), die gekenmerkt worden door verhoogde niveaus van autofagie in vergelijking met meer gedifferentieerde kankercelpopulaties, zoals waargenomen in meerdere kankertypes, waaronder blaas- en borstkanker [112,113]. Deze hoge niveaus van autofagie zijn essentieel om CSC-eigenschappen zoals rust te behouden.
Autofagie speelt een regulerende rol tijdens de aanpassing van kankercellen aan hypoxische stress in slecht geoxideerde gebieden van solide tumoren of in het gebied van het beenmerg na infiltratie van acute myeloïde leukemie [114].
Bovendien stelt het begin van autofagie kankercellen in staat zich te beschermen tegen de werking van chemotherapeutische toxines, waardoor het apoptotische effect dat wordt veroorzaakt door medicamenteuze behandeling wordt geblokkeerd (autofagie-gemedieerde resistentie) [115]. Bijvoorbeeld, cisplatina-resistente ovariumkankercellen worden gekenmerkt door verhoogde niveaus van autofagische flux [116].
Tot slot zijn er geneesmiddelen die in staat zijn autofagische celdood te induceren en die worden gebruikt om een gro
ter therapeutisch resultaat te bereiken bij apoptosis-resistente tumorcellen [117,118].
De co-existentie van verschillende cellulaire en moleculaire processen binnen een tumormassa heeft het onderzoek geleid naar de identificatie van sleutelmoleculen in de autofagie/apoptose-schakelaar (Beclin1, Caspase, p53, PI3K/AKT/mTOR en p62), die nuttig is voor het plannen van anti-kanker doeltherapieën [119,120].
Preklinische studies hebben het belang van het onderdrukken van autofagie in therapeutische strategieën benadrukt. KRAS is een gen dat celproliferatie controleert; als het gemuteerd is, blijven de cellen zich prolifereren en ontwikkelen ze zich tot kanker. BRAF is een gen dat codeert voor een eiwit bekend als serine/threonine-proteïnekinase B-Raf. Dit eiwit is betrokken bij de activering van cellulaire signalen naar celproliferatie; in veel menselijke tumoren is dit gen defect [121,122]. In menselijke cellijnen met dergelijke mutaties werden hoge niveaus van autofagie waargenomen. KRAS- en Tp53-status is ook zeer belangrijk; inderdaad, bij KRAS-gemuteerde muizen die de genen ATG5 of ATG7 missen, werden pre-maligne laesies in de alvleesklier onthuld, terwijl bij muizen die KRAS en Tp53 missen, het verlies van de genen ATG5 of ATG7 leidde tot de verwerving van kwaadaardigheid en de ontwikkeling van adenocarcinomen [123]. Deze resultaten suggereren dat remming van autofagie in BRAF- of KRAS-mutaties in tumoren een potentieel antikankerdoelwit zou kunnen zijn.
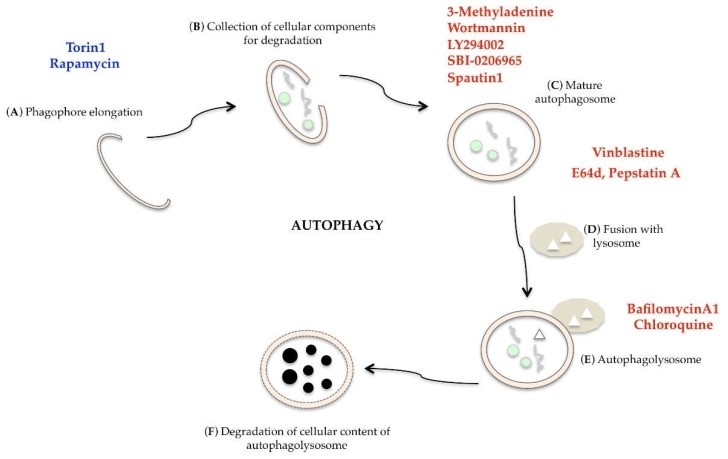
In de afgelopen jaren zijn veel verbindingen geïdentificeerd die autofagie kunnen induceren of remmen voor farmacologische interventie, maar hun gebruik moet breed worden ondersteund door preklinische gegevens [124]. Torin1, een krachtige en selectieve remmer van mTOR, en rapamycine, een mTORC1-remmer, kunnen autofagie stimuleren [125]. Autofagie wordt op meerdere niveaus geremd door 3-methyladenine, LY294002, wortmannine (PI3K-specifieke remmers), SBI-0206965 (ULK1-remmer), spautin1 (remmer van UB-specifieke peptidase), vinblastine (blokkeert de fusie tussen lysosoom en autofagosoom), E64d en pestatin A (remmers van lysosomale proteasen), bafilomycine A1 (remmer van ATPase-pomp) en chloroquine (lysosomotroop middel dat interferentie veroorzaakt met de afbraak van autofagosomen) (Figuur 9) [124,126].
Hydroxychloroquine, het antimalariaderivaat van chloroquine, is uitgeprobeerd in klinische onderzoeken (Tabel 3); het is in staat de lysosomale enzymdegradatieve werking binnen het autofagolysosoom te blokkeren. Hydroxychloroquine wordt in plaats van chloroquine gebruikt omdat de remmende werking op autofagie minder giftig is. Veel klinische studies hebben de effectiviteit ervan aangetoond wanneer het alleen of in combinatie met andere chemotherapeutica wordt toegediend, en andere studies hebben aangetoond dat het effectief is tegen verschillende solide tumoren, zoals glioblastoma, astrocytoom, long- en alvleesklierkanker [10].
Tabel 3
Lijst met enkele klinische onderzoeken van hydroxychloroquine.
| Combinatie van Behandelingstype | Type tumor | Klinisch Onderzoek op ClinicalTrials.gov |
|---|---|---|
| Sirolimus of vorinostat + HCQ | Geavanceerde solide tumoren | NCT01266057 |
| Enkele HCQ | Glioblastoma en astrocytoom | NCT02432417 |
| HCQ + Vorinostat | Maligne solide neoplasma | NCT01023737 |
| Cisplatine, etoposide + HCQ | Stadium 4 kleincellige longkanker | NCT00969306 |
| HCQ + Abraxane + Gemcitabine | Pancreatische adenocarcinoma | NCT01978184 |
| Sorafenib + HCQ | Refractaire of teruggekeerde solide tumoren | NCT01634893 |
Bovendien hebben veel natuurlijke producten uit levende organismen biologische en farmacologische eigenschappen die autofagie moduleren; deze potentiële toepassingen kunnen worden bestudeerd op in vitro modellen en vertaald naar klinische toepassing [127].
10. Autofagie en Auto-immuunziekten
Auto-immuunziekten, zoals systemische lupus erythematosus (SLE), worden gekenmerkt door significante veranderingen in de prognose en uitkomst van de behandelingen. Ze omvatten het gehele immuunsysteem, inclusief B- en T-cellen van het adaptieve systeem, en dendritische cellen, macrofagen en neutrofielen van het aangeboren systeem, met als gevolg de productie van antinucleaire lichamen. Metabole paden zijn belangrijke regulatoren van de differentiatie en activatie van het immuunsysteem, dus als ze verstoord zijn, kunnen ze leiden tot de ontwikkeling van auto-immuunziekten [128]. Metabole controle van de differentiatie van het immuunsysteem begint bij hematopoëtische stamcellen, die mTOR-afhankelijke autofagie activeren [129]. mTOR is een voedingsstof-sensitieve kinase die, door metabole signalen te koppelen aan genetische patronen, celgroei en differentiatie controleert. In het bijzonder vormt mTOR twee complexen: mTORC1, dat Th1- en Th17-ontwikkeling stimuleert, en mTORC2, dat Th2-ontwikkeling bemiddelt; beide complexen beperken de ontwikkeling van regulerende T-cellen (Treg) [130]. Tijdens metabole stress bevordert activatie van endosomaal verkeer door Rab4A autofagie via CD4- en CD3-oppervlaktereceptoren en de dynamin-gerelateerde eiwit 1 (Drp1) mitochondriale fissie initiator. Lysosomale afbraak van Drp1 vermindert mitofagie, met als gevolg ophoping van grote langwerpige mitochondriën en ROS-generatie in lupus T-cellen [131]. ROS activeert mTORC1, wat leidt tot pro-inflammatoire necrotische dood en IL-4- en IL-17-secretie door DN T-cellen en uitputting van CD8-geheugen T-cellen en Treg-cellen [132,133]. In de lever gaat deze activering vooraf aan de productie van antinucleaire antilichamen en het begin van de ziekte bij lupusgevoelige muizen [134]. Deze gegevens tonen een pathogene rol voor Rab4-gemedieerde Drp1-depletie en identificeren het als potentieel doelwit voor SLE-behandeling [131]. Hoewel autofagie uitgebreid is onderzocht in SLE T-cellen, is autofagie in B-cellen minder bestudeerd. Clarke en medeauteurs tonen een verhoogde autofagie aan in muizen- en menselijke lupus B-cellen en dat deze nodig is voor de ontwikkeling van plasmablasten [135]. Ze toonden de belangrijke rol van autofagie aan in de differentiatie van plasmablasten, omdat B-cellen geïsoleerd uit muizen met ATG7-deficiëntie na in vitro stimulatie niet effectief differentieerden tot plasmacellen. Op dezelfde manier differentieerden gestimuleerde menselijke B-cellen na remming van autofagie niet tot plasmablasten. Daarom ontdekten ze, vóór het begin van de ziekte, in een vroeg stadium van B-cellen van het SLE-muismodel, een activering van autofagie die toenam met de leeftijd [135].
Het onevenwicht tussen perifere Th17- en Treg-cellen is cruciaal voor de pathogenese van SLE en andere auto-immuunziekten [136]. Th17-cellen, die IL-17A, IL-17F en IL-22 produceren, bevorderen de auto-immuunrespons en ondersteunen de productie van auto-antilichamen door B-cellen [137]. Treg-cellen reguleren de immuunhomeostase door IL-10 en TGF-β vrij te geven. Het belangrijkste Treg-specifieke transcriptiefactor is de forkhead-box-eiwit 3 (Foxp3); de afname van Foxp3-expressie in Treg-cellen is verantwoordelijk voor de disfunctie van Treg in SLE [138]. Gezien de cruciale rol van autofagie in de metabole checkpoints en de controle van auto-immuniteit, met name in de pathogenese van SLE, hebben recente studies zich gericht op autofagie-gerichte therapieën.
Sommige studies hebben gemeld dat HCQ een hoeksteen is van SLE en andere immuunziekten, zoals de behandeling van reumatoïde artritis [139]. Zowel Th17- als Treg-cellen vertonen autofagieactivatie bij SLE, en door het autofagiepad te blokkeren met CQ, zou de Th17/Treg-respons kunnen worden hersteld. Zo vermindert remming van autofagie de IL-17-secretie door de overactiviteit van Th17-cellen die wordt waargenomen bij SLE en verhoogt het de expressie van Foxp3 in Treg-cellen, waarbij de functionaliteit van Treg-cellen behouden blijft. Hierdoor wordt het immuunevenwicht hersteld, wat resulteert in lagere serumniveaus van inflammatoire cytokines en auto-antilichamen [140].
Patiënten met SLE hebben Treg-disfunctie als gevolg van mTOR-activatie. Rapamycine, werkend op mTOR, remt de proliferatie van T-cellen en is ontwikkeld als medicatie onder de generieke aanduiding sirolimus [141]. In vivo-onderzoek toonde aan dat sirolimus auto-immuniteit bij lupusgevoelige muizen teniet deed [142]. Een recent prospectief onderzoek toonde de werkzaamheid en veiligheid van sirolimus aan bij SLE-patiënten met ernstige en aanhoudende ziekte [143].
Aangezien SLE kan worden geassocieerd met een verhoogd risico op maligniteit, voornamelijk lymfomen, kan de behandeling met immunosuppressieve geneesmiddelen ook bijdragen aan de ontwikkeling van secundaire kankers [144]. Opkomende gegevens suggereren dat rapamycine een effectief antineoplastisch middel is bij leukemie en lymfoom, en daarom zou het kunnen worden gebruikt als een immunosuppressief middel voor de behandeling van SLE-patiënten [145]. Het is echter niet altijd duidelijk of farmacologische modulatie van autofagie positieve of negatieve effecten heeft; daarom moeten meer gedetailleerde studies worden uitgevoerd om een specifieke doelgerichte therapie te ontwikkelen.
11. Conclusies
Autofagie is een sterk geconserveerd afbraakproces doorheen de evolutie. Veel Nobelprijzen zijn toegekend aan onderzoekers die hun aandacht hebben gericht op de studie en het begrip van dit fenomeen: in 1974 beschreef Dr. Christian de Duve autofagie als een proces van "zelfeten", en in 2016 werd Dr. Yoshinori Ohsumi erkend voor de opmerkelijke bijdrage aan de identificatie van essentiële genen voor autofagie [146,147].
Veel studies hebben aangetoond dat autofagie een fundamentele rol speelt in het handhaven van de cellulaire energetische en fysiologische balans; daarom behoort verstoring van dit fenomeen tot de oorzaken van steeds frequenter voorkomende ziekten. Veel farmacologische behandelingen en passende levensstijlen zijn gericht op het herstellen van de fysiologische niveaus van autofagie, wat suggereert dat dit proces een potentieel en veelbelovend doelwit is van openbaar en gezondheidsbelang.
Afkortingen
ATG Autofagie
mTOR Mammalian target of rapamycin
PKA Proteïnekinase A
ULK1 Unc-51-like kinase 1
NAD+ Nicotinamide-adeninedinucleotide
AMPK AMP-geactiveerde proteïnekinase
FIP200 Focal adhesion kinase family interacting protein of 200 kDa
PtdIns3K Fosfatidylinositol 3-kinase
VPS Vacuolaire-eiwitsorteerder
AMBRA1 Activerend molecuul in BECN1-gereguleerde autofagie-eiwit 1
UVRAG Ultraviolette straling resistente geassocieerd gen
PI3P Fosfatidylinositol-3-fosfaat
WIPI WD-herhalend eiwit dat interageert met fosfoïnositiden
MAP1-LC3 Microtubule-associated protein 1 light chain 3
PE Fosfatidylethanolamine
NBR1 Buur van BRCA1-gen 1
NDP52 Nucleair domein 10-eiwit 52
UB Polyubiquitineketting
UBD UB-bindend domein
LIR LC3-interagerende regio's
SQSTM1 Sequestosome 1
BNIP3 Bcl-2/adenovirus E1B 19-kDa interagerend eiwit 3
NLR (NOD)-achtige receptor
IL Interleukine
IGF-1 Insulineachtige groeifactor 1
Sirt-1 Sirtuïne-1
HDL High density lipoprotein
EGFR Epidermale groeifactor receptor
HSP70 Heat shock proteïne 70
TPD-43 TAR DNA-bindend eiwit 43
PS1 Preseniline 1
Nrf2 Kernfactor afgeleid van erytroïde 2-achtige 2
TFEB EB-transcriptiefactor
LRRK2 Leucinerijke herhalingskinase 2
PARK2 Parkin RBR E3-ubiquitine-proteïneligase
PINK1 PTEN-geïnduceerde veronderstelde kinase 1
HTT Huntingtine
CSC Kankerstamcellen
SLE Systemische lupus erythematosus
Treg Regulerende T-cellen
Drp1 Dynamin-gerelateerd eiwit 1
Foxp3 Forkhead-box-eiwit 3

